UNIVERSITA’ DEGLI STUDI DI TORINO
Dipartimento di Genetica, Biologia e Biochimica
DOTTORATO DI RICERCA IN BIOCHIMICA E BIOTECNOLOGIA CELLULARE
CICLO XVI
TITOLO DELLA TESI
Parte prima
"EFFETTO SINERGICO DELL’ACIDO RETINOICO E DEL DEIDROEPIANDROSTERONE SUL DIFFERENZIAMENTO DI CELLULE DI NEUROBLASTOMA UMANO"
Tesi presentata da:
Dott. Vincenzo Guarnieri
Tutor:
Prof: Gian Piero Pescarmona
Coordinatore del ciclo:
Prof. Amalia Bosia
Prof. Dario Ghigo
Anni Accademici: 2000-2004
INDICE GENERALE
PREFAZIONE....................................................................................................1
PARTE PRIMA
EFFETTO SINERGICO DELL’ACIDO RETINOICO E DEL DEIDROEPIANDROSTERONE SUL DIFFERENZIAMENTO DI CELLULE DI NEUROBLASTOMA UMANO
CAPITOLO 1 - INTRODUZIONE.................................................................4
L’ACIDO RETINOICO E I RETINOIDI...................................................................4
Generalità...................................................................................................4
Origine dei retinoidi...................................................................................5
Meccanismo d’azione dei retinoidi............................................................6
Funzioni dell’acido retinoico e dei recettori nucleari...............................9
IL DEIDROEPIANDROSTERONE.......................................................................14
Generalità.................................................................................................14
Biosintesi del DHEA.................................................................................14
Proprietà biologiche del DHEA...............................................................17
DHEA e tumore........................................................................................18
DHEA e sistema nervoso centrale............................................................18
Meccanismi d’azione del DHEA..............................................................20
LE METALLOPROTEASI..................................................................................25
LE TRANSGLUTAMINASI................................................................................27
SCOPO DEL LAVORO......................................................................................30
CAPITOLO 2 - PROCEDURE SPERIMENTALI.....................................31
COLTURE CELLULARI....................................................................................31
PROLIFERAZIONE CELLULARE........................................................................31
MOVIMENTO E CRESCITA DEI NEURITI...........................................................31
IMMUNOFLUORESCENZA................................................................................32
WESTERN BLOT.............................................................................................32
ANALISI ZIMOGRAFICA..................................................................................33
BIOTINILAZIONE DELLA SUPERFICIE CELLULARE...........................................33
ANALISI STATISTICA......................................................................................34
CAPITOLO 3 - RISULTATI........................................................................35
RA E DHEA INIBISCONO LA PROLIFERAZIONE CELLULARE..........................35
EFFETTO DI RA E DHEA SULLA MOTILITÀ CELLULARE................................36
RA E DHEA SINERGIZZANO NEL PROMUOVERE IL DIFFERENZIAMENTO CELLULARE....................................................................................................37
L’AZIONE SINERGICA DI RA E DHEA SUL DIFFERENZIAMENTO DEL NEUROBLASTOMA NON È MEDIATA DA UN’ALTERAZIONE DELL’ESPRESSIONE DEI RECETTORI PER L’ACIDO RETINOICO........................................................40
MODULAZIONE DELL’ATTIVITÀ METALLOPROTEASICA DURANTE IL DIFFERENZIAMENTO......................................................................................40
ESPRESSIONE DELLA TRANSGLUTAMINASI.....................................................42
CAPITOLO 4 - DISCUSSIONE...................................................................44
Prefazione
Il filo conduttore degli studi svolti durante il Dottorato di Ricerca in Biochimica e Biotecnologia Cellulare è stato l’analisi dei meccanismi coinvolti nel differenziamento cellulare. Il passaggio da stato proliferativo a stato differenziato è un fenomeno complesso, che prevede lo spegnimento di alcune vie metaboliche e l’attivazione di altre, al fine di specializzare la cellula da un punto di vista strutturale e metabolico.
Nel corso dei quattro anni mi sono principalmente occupato di due progetti che hanno toccato aspetti diversi del differenziamento cellulare.
Il primo è stato svolto durante i primi due anni presso il laboratorio di biochimica del prof. G.P. Pescarmona del Dipartimento di Genetica, Biologia e Biochimica dell’Università degli Studi di Torino. L’oggetto di studio di questo progetto è stato il differenziamento di una linea umana di neuroblastoma. Su questo modello tumorale si è voluto analizzare il noto effetto differenziante dell’acido retinoico, in presenza di deidroepiandrosterone (DHEA), uno steroide dalle riconosciute proprietà antiproliferative e chemopreventive, e ancora oggetto di molti studi poichè il suo meccanismo d’azione rimane tuttora incerto.
Il secondo progetto è iniziato nel corso del terzo anno, durante il periodo svolto nell’ambito dell’Exchange Visitor Program of the United States Department of State presso il laboratorio della Prof.ssa Bonnie Firestein (Neuroscience and Cell Biology Department, Rutgers University, Piscataway, New Jersey). Si è trattato dell’identificazione di binding partners dell’enzima nNOSμ. Questa isoforma enzimatica compare nel corso del differenziamento delle cellule muscolari scheletriche, e il significato di tale induzione rimane da chiarire. Lo studio è poi proseguito nel quarto anno presso il laboratorio di Biochimica del CeRMS (Centro Ricerche in Medicina Sperimentale), Ospedale San Giovanni Battista, Torino, e presso il Dipartimento di Genetica, Biologia e Biochimica, dell’Università degli Studi di Torino. Durante quest’ultimo anno mi sono in particolare occupato dell’interazione tra la nNOSμ e la H-FABP (Heart type-Fatty Acid Binding Protein), una proteina altamente espressa nel muscolo differenziato. Di conseguenza la mia tesi risulta suddivisa in due parti indipendenti.
PARTE PRIMA
"EFFETTO SINERGICO DELL’ACIDO RETINOICO E DEL DEIDROEPIANDROSTERONE SUL DIFFERENZIAMENTO DI CELLULE DI NEUROBLASTOMA UMANO"
Capitolo 1
INTRODUZIONE
L’acido retinoico e i retinoidi
Generalità
Gli organismi multicellulari richiedono l’esistenza di specifici meccanismi di comunicazione intercellulare per organizzare la loro complessa struttura durante l’embriogenesi e per mantenere le funzioni fisiologiche di ogni organo durante la vita.
L’acido retinoico (RA) e in generale i retinoidi sono molecole segnale che, insieme ai loro recettori nucleari, stabiliscono dei networks di comunicazione genetica essenziali per lo sviluppo embrionale e per il mantenimento dello stato differenziato dei tessuti attraverso la regolazione della proliferazione, differenziamento e morte cellulare (4-6). Di particolare importanza è il ruolo esercitato sullo sviluppo e sulle funzioni fisiologiche del sistema nervoso e del sistema riproduttivo (5).
I retinoidi, grazie al loro potere di condurre al differenziamento cellulare, offrono grandi potenzialità nella terapia del cancro. Gli effetti benefici della "terapia differenziante" sono in particolare ben conosciuti nel caso della leucemia acuta promielocitica, il cui trattamento standard prevede la combinazione di all-trans RA e di chemioterapia. Non bisogna tuttavia trascurare il fatto che in certe condizioni i retinoidi siano dei potenti teratogeni (61). E’ quindi indispensabile conoscere in modo approfondito le loro funzioni per poterne fare un corretto impiego. 4 Dottorato di Ricerca in
Origine dei retinoidi
I retinoidi derivano dalla vitamina A (retinolo) introdotta con la dieta, in particolare con uova, latte, burro e olio di fegato di pesce, e dal β-carotene di origine vegetale. Dopo uptake ad opera delle cellule della mucosa intestinale, il retinolo viene esterificato e il retinil estere formatosi passa nel flusso sanguigno sottoforma di VLDL (very low density lipoprotein) da cui viene prelevato e immagazzinato dal fegato. In seguito alla rimozione attraverso il clivaggio del retinil estere, il retinolo viene ossidato a retinale e quindi ad acido retinoico (vedi fig. 1).
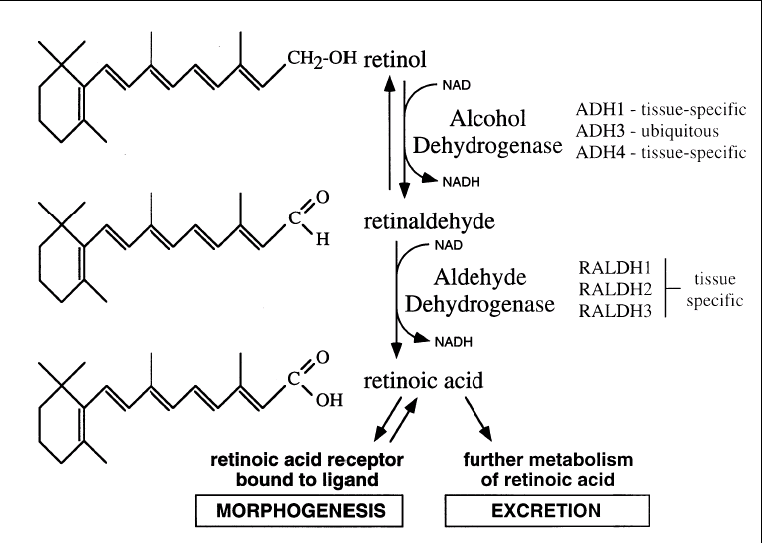
Fig 1. Biosintesi dell’acido retinoico da retinolo
Il primo step viene catalizzato da alcool deidrogenasi (ADH), in particolare da tre classi: ADH1, ADH2 e ADH3. La più attiva ed importante è la ADH3 che viene espressa ubiquitariamente nell’organismo.
La retinaldeide deidrogenasi (RALDH) catalizza il secondo step. Anche questo enzima è presente in tre classi diverse (RALDH1, RALDH2 e RALDH3) che vengono espresse in maniera tessuto-specifica. Solo i tessuti in grado di esprimere questo enzima possono produrre acido retinoico. Si noti che sia ADH che RALDH richiedono NAD+ per il loro funzionamento. Inoltre il secondo step è irreversibile e quindi molto delicato dal momento che, una volta sintetizzato, l’acido retinoico non può più essere riconvertito in retinolo o retinale (62).
RALDH è un enzima chiave nello sviluppo embrionale. Esperimenti dimostrano che la sua assenza causa una precoce morte embrionale.
E’ interessante notare che solo una piccola porzione (0.2-5%) del retinolo presente nel plasma o nei tessuti viene convertita in acido retinoico all-trans (ATRA), il principale attivatore dei recettori per l’acido retinoico. Altri metaboliti con funzione di molecola segnale sono l’acido 9-cis RA e 13-cis RA. Quest’ultimo ha un’attività inferiore rispetto agli altri due (61).
Meccanismo d’azione dei retinoidi
L’azione dei retinoidi si esplica attraverso due tipi di recettori nucleari appartenenti alla famiglia dei recettori degli ormoni steroidei: RAR (Retinoic Acid Receptor) e RXR (Rexinoic Receptor) (7,8). Entrambi sono espressi in almeno tre isoforme denominate con α, β e γ, che differiscono nella distribuzione spazio-temporale e nell’affinità dei ligandi (9-11).
I RAR e gli RXR sono fattori di trascrizione che funzionano principalmente come RAR-RXR eterodimeri. Questi eterodimeri presentano due funzioni distinte. La prima è quella di modulare la trascrizione di geni target in seguito a binding con i RARE (Retinoic Acid Response Element) presenti nei promoters di tali geni. La seconda consiste nel modificare l’efficienza di altri pathways attraverso meccanismi non ancora del tutto chiari. L’effetto di questo tipo più importante è senz’altro la repressione dell’attivazione trascrizionale mediata da
AP-1. E’ infatti anche grazie a questa inibizione che si può spiegare l’effetto anti-oncogenico dell’acido retinoico (61).
L’isomero all-trans RA è il principale ligando e attivatore di RAR ma non di RXR. Il 9-cis RA invece è in grado di legare e attivare sia RAR che RXR. Non è stato però ancora dimostrato che tale molecola sia il ligando fisiologico di RXR dal momento che sono stati identificati altri possibili ligandi endogeni (genericamente denominati "rexinoids").
In presenza di antagonisti del dimero RAR-RXR o in assenza dei loro ligandi, i geni target sono repressi. Questo può essere possibile a causa del reclutamento di HDAC (Histone deacetylase-containing complex) legato per mezzo di corepressori (CoR) all’eterodimero (apo-) RAR-RXR (vedi figura 2, in alto). I due corepressori più importanti sono NCoR (nuclear receptor corepressor) e SMRT (silencing mediator for retinoid and tyroid hormone receptor). In questa situazione si verifica una deacetilazione degli istoni, una condensazione della cromatina e il conseguente silenziamento del gene.
Il binding di un agonista di RAR destabilizza il legame con il CoR e induce delle modificazioni allosteriche nella regione LBD (ligand binding domain). Tali modificazioni generano una nuova superficie di interazione che, in un primo momento, permette la formazione di un complesso costituito dall’eterodimero (holo-) RAR-RXR e da un coattivatore (CoA). Il CoA lega HAT (histone acetyltransferase) che permette la decondensazione della cromatina (fig 2, parte centrale). Successivamente si forma un complesso con TRAP (thyroid hormone receptor-associated protein) o con DRIP (vitamin D receptor-interacting protein) o con SMCC (Srb and mediator protein –containing complexes), il quale lega i macchinari per la trascrizione che può così avvenire (fig 2, in basso) (61).
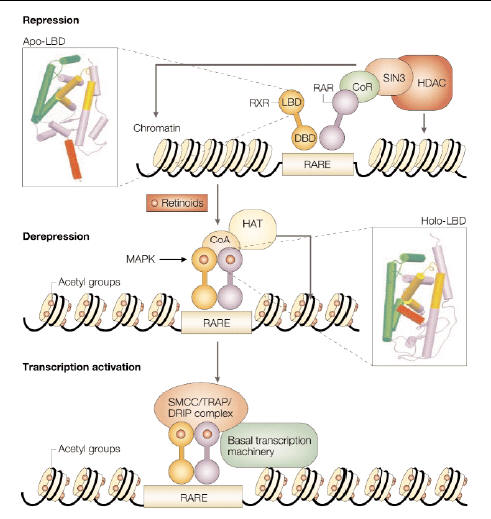
Fig 2. Meccanismi di repressione e attivazione trascrizionale attraverso l’eterodimero RAR-RXR.
Funzioni dell’acido retinoico e dei recettori nucleari
Come precedentemente detto, RA è essenziale nella regolazione dell’embriogenesi. Una bassa concentrazione di vitamina A causa malformazioni, mentre se la concentrazione è elevata si hanno effetti teratogeni. Questo supporta l’idea che RA sia un agente importante per la morfogenesi. Alcuni esperimenti mostrano che quasi tutte le malformazioni congenite causate da deficienza di vitamina A sono dovute alla assenza delle funzioni di RAR e RXR. Quindi una dose sub-teratogenica di RA può prevenire alcuni difetti congeniti, indicandone una notevole importanza nella gravidanza. Nell’adulto i retinoidi sono indispensabili per il corretto funzionamento di diversi organi tra cui fegato, pelle, polmoni, sistema nervoso e sistema immunitario.
Per quel che riguarda l’attività antitumorale dei retinoidi, bisogna considerare che esistono molti fattori che possono contribuire alla nascita di un tumore come ad esempio le modificazioni geniche o la carcinogenesi chimica. I retinoidi possono interferire con questi fattori su vari livelli (vedi fig 3).
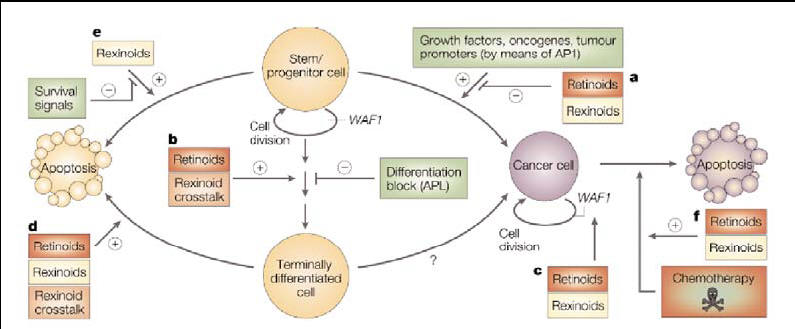
Fig 3. Punti chiave in cui i retinoidi e i rexinoidi possono interferire sulla genesi tumorale. (a) blocco della carcinogenesi chimica probabilmente grazie all’inibizionie di AP-1. (b) induzione e stabilizzazione del differenziamento. (c) blocco del ciclo cellulare della cellula cancerogena in G1. (d) effetto apoptotico post-maturazione. (e) morte delle cellule premature ad opera solo dei "rexinoids" in certe condizioni. (f) induzione di apoptosi di cellule cancerogene (senza o in combinazione con agenti chemioterapici)
Le azioni principalmente note consistono nell’induzione del differenziamento e/o apoptosi di cellule tumorali e nell’inibizione della propagazione tumorale nei casi di cancro chimicamente indotti.
La tabella 1 riassume le applicazioni cliniche dei retinoidi.
Analizzando più nello specifico i recettori nucleari che mediano l’effetto di RA si nota che una disfunzione di RARα è alla base della leucemia mieloide. Infatti questa forma tumorale è causata da una traslocazione genica che porta all’espressione di una proteina di fusione contenente RARα. In questo modo il recettore pur mantenendo la capacità di accettare il ligando e di legarsi al DNA riconoscendo i RARE, presenta una regolazione alterata. Infatti rispetto a RARα wild type, si ha una maggior affinità con HDAC (e con i corepressori) e quindi affinchè si verifichi una dissociazione di tale complesso sono necessarie elevate (e non fisiologiche) concentrazioni di acido retinoico (61). La terapia con RA nella leucemia mieloide rappresenta uno dei rari casi di successo nella lotta contro il cancro. In generale RARα esplica un ruolo critico nel mediare l’effetto inibitorio sulla proliferazione che i retinoidi manifestano su molte cellule cancerogene. La sua espressione è inoltre indotta dagli estrogeni nelle cellule di carcinoma mammario (16,17).
RARβ viene considerato in molti lavori un soppressore tumorale, coinvolto in particolare in un vasto range di tumori solidi (tabella 2).
La perdita delle funzioni di RARβ comporta la progressione di tumore nell’uomo. Conseguentemente, la riattivazione del recettore genera un effetto opposto. Ciò è stato osservato introducendo RARβ ricombinante esogeno in linee di cellule tumorali non responsive a RA. Molto interessante, nelle linee di carcinoma mammario sembra che il promoter del gene di RARβ sia silente. Questo può essere revertito attraverso demetilazione del DNA o inibizione di HDAC.
Tabella 1. Effetti clinici dei retinoidi
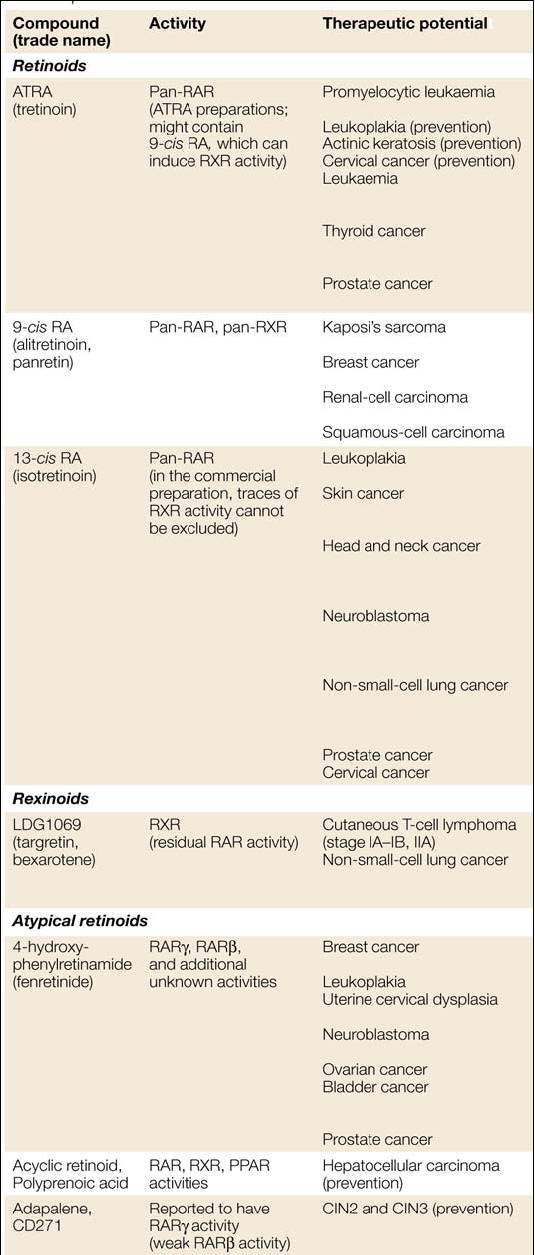
Tabella 2. Coinvolgimento dei RAR/RXR nella progressione tumorale
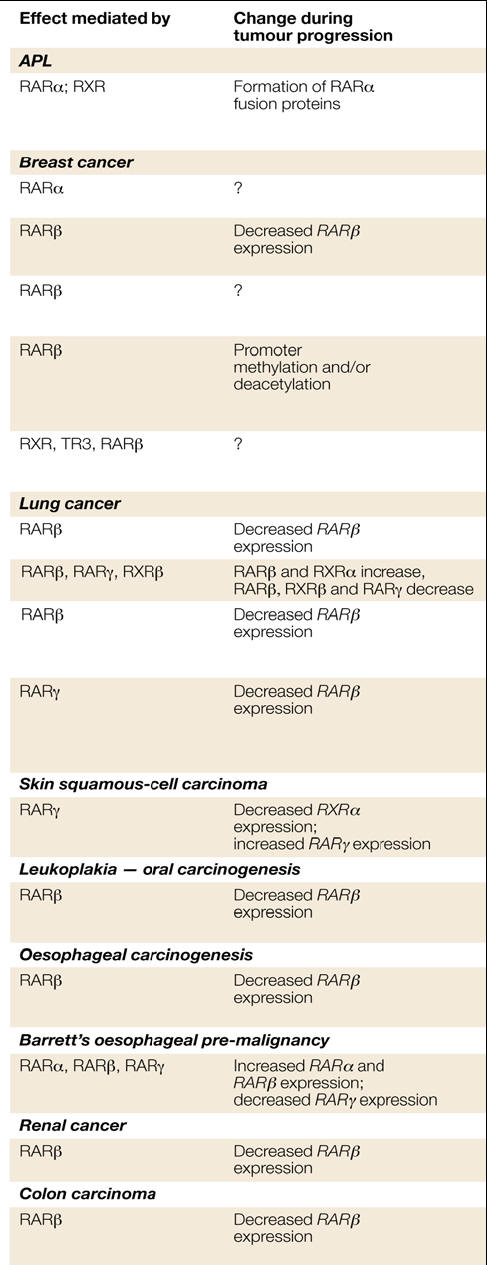
RARβ possiede delle caratteristiche che lo distinguono dagli altri recettori. Innanzitutto, interagisce più debolmente con i corepressori, di conseguenza l’eterodimero RARβ-RXR risulta più sensibile all’attivazione ad opera dei retinoidi. In secondo luogo, RARβ inibisce costitutivamente AP1 in modo ligando-indipendente. Per queste caratteristiche RARβ sfavorisce una rapida crescita tumorale (12-15).
Gli RXR sono comunemente visti come partners di eterodimerizzazione dei RAR. La loro presenza permette o amplifica gli effetti dei retinoidi mediati dai RAR. Recentemente gli RXR vengono anche considerati come mediatori attivi dei segnali di soppressione tumorale, indipendentemente dagli altri recettori nucleari. Sono stati infatti osservati effetti come la prevenzione e la regressione del carcinoma mammario con segni di adipogenesi, attribuibili solo al ruolo di RXR. Inoltre sembra che i ligandi di questi recettori non presentino gli effetti secondari normalmente associati alla terapia con RA.
Bisogna sottolineare che i recettori RXR sono in grado di eterodimerizzare con tutti gli altri fattori nucleari appartenenti alla famiglia degli steroidi, come il recettore della vitamina D, dell’ormone tiroideo e degli estrogeni (18-20).
Il Deidroepiandrosterone
Generalità
Il deidroepiandrosterone (DHEA) e il suo estere solfato (DHEAS) sono ormoni steroidei al centro da tempo di infinite discussioni. Questi ormoni sono stati ampiamente pubblicizzati e hanno attratto l’attenzione della letteratura scientifica per il loro presunto effetto "anti-invecchiamento", al punto che numerosi studiosi ritengono che i livelli plasmatici di DHEA e DHEAS possano essere considerati dei validi indicatori di invecchiamento fisiologico. Il grande interesse rivolto nei confronti del DHEA(S) deriva dal fatto che i suoi livelli plasmatici mostrano una significativa riduzione con il progredire dell’età, il che fa supporre un suo possibile coinvolgimento nei processi di invecchiamento. Malgrado la scoperta del DHEA e del DHEAS risalga a più di cinquanta anni fa, il loro significato biologico e meccanismo d’azione sono attualmente ancora oggetto di numerose controversie.
Biosintesi del DHEA
Nell’uomo tutti gli ormoni steroidei derivano dal DHEA che a sua volta viene prodotto a partire dal colesterolo.
La sintesi del DHEA avviene principalmente a livello della zona corticale delle cellule reticolari del surrene, sotto il controllo dell’ormone ipofisario adrenocorticotropo ACTH (63). Minori quantità di DHEA sono prodotte dal testicolo e dall'ovaio (64); questo steroide è inoltre anche prodotto e metabolizzato a livello del cervello, in particolare dalle cellule gliali, dove è presente in concentrazione molto elevata e sembra svolgere la funzione di neurosteroide (65).
La sintesi del DHEA richiede la rimozione della catena laterale in C-17 dell’anello D del colesterolo. La rimozione ha luogo nei mitocondri dei tessuti in cui l’ormone è prodotto e richiede l’introduzione di due gruppi ossidrili in due atomi di carbonio adiacenti alla catena laterale (C-20 e C-22) e la successiva rottura del legame che li unisce con formazione di Δ5-pregnenolone e una aldeide a sei atomi di carbonio, l’aldeide isocaproica. Il Δ5-pregnenolone è l’intermedio comune nella biosintesi di tutti gli ormoni steroidei.
Nelle tappe successive che riguardano la biosintesi del deidroepiandrosterone, il Δ5-pregnenolone viene convertito dalla 17-α-idrossilasi del reticolo endoplasmatico in 17-α-idrossipregnenolone che, in seguito alla rimozione della catena laterale di due unità carboniose da parte di una liasi 17-20, viene convertito in deidroepiandrosterone. (figura 4).
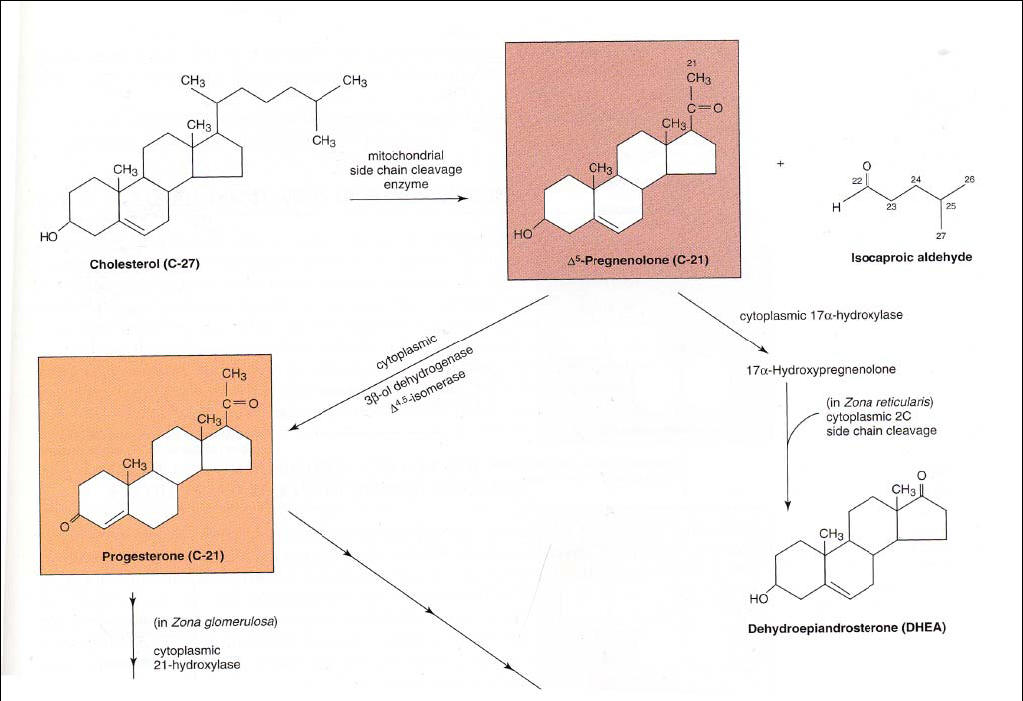
Fig 4. Biosintesi del deidroepiandrosterone
Il DHEA è presente nell’organismo sia in forma libera, sia in forma coniugata con acido glucuronico o solfato (DHEAS) (figura 5). Le due forme DHEA e DHEAS sono interconvertibili tra loro grazie all’intervento di due enzimi, una solfotransferasi e una solfatasi.
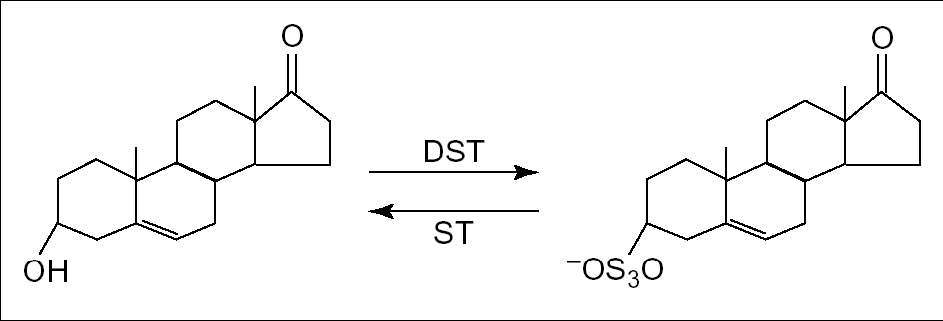
Fig 5. Struttura chimica del deidroepiandrosterone (DHEA) e il suo estere solfato (DHEAS) DST: solfotransferasi. ST: solfatasi
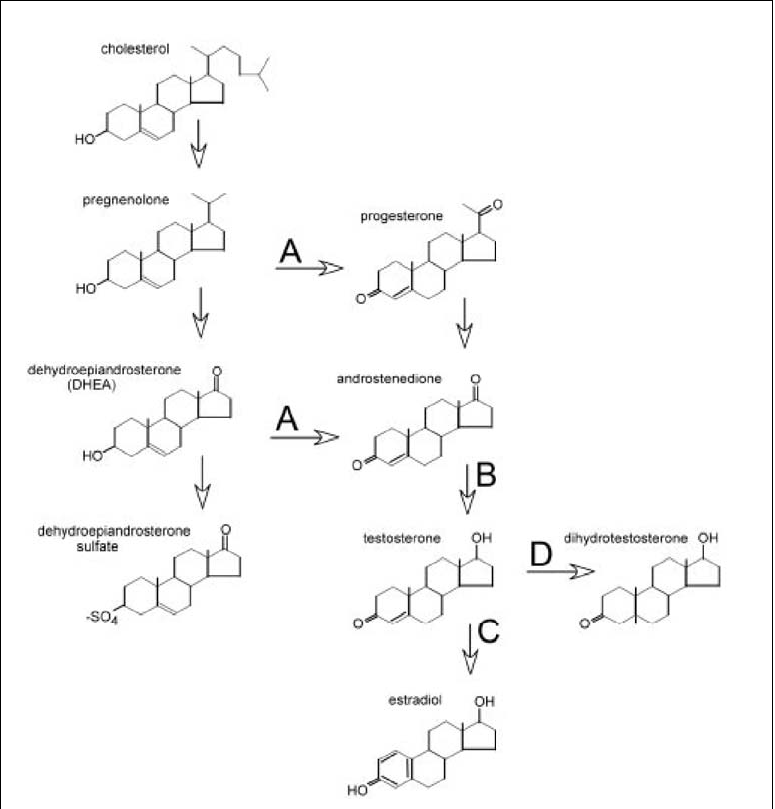
Fig 6. Il DHEA come precursore degli ormoni sessuali
La forma solfatata deriva esclusivamente dal surrene in quanto il processo di solfatazione avviene a livello di tale ghiandola.
A livello dei tessuti periferici è espressa una solfatasi che converte il DHEAS in DHEA, che può essere in seguito trasformato nelle forme attive degli steroidi sessuali, estradiolo e testosterone; nella figura 6 sono riportate le tappe enzimatiche responsabili della trasformazione del DHEA nei vari steroidi sessuali.
L’estere solfato DHEAS è la forma più abbondante nel circolo sanguigno ed è presente ad una concentrazione che oscilla tra 0,2-5 μg/ml nei soggetti giovani, rispetto al DHEA che non supera i 5 ng/ml.
Nel plasma circa l’80% del DHEA (nella forma solfatata) è legato all’albumina e il restante 20% è legato a lipoproteine nella forma DHEA-Fatty Acid (66): il legame a tali proteine permette di mantenere costante la concentrazione di questo steroide nel circolo ematico e di proteggerlo dal catabolismo e dall’inattivazione.
L’inattivazione dello steroide avviene a livello epatico ad opera di reazioni di riduzione dell’anello ciclopentanoperidrofenatrenico: l’ormone è reso in questo modo più solubile e facilmente coniugabile con glucuronidi o solfato ed eliminato successivamente con le urine.
Proprietà biologiche del DHEA
Il contenuto dello steroide nell’organismo è strettamente correlato all’età; nelle donne in gravidanza il DHEA viene prodotto anche dalla placenta e il feto stesso lo sintetizza a partire dal secondo trimestre di vita. La presenza di questo ormone sembra quindi essere indispensabile per un corretto sviluppo del feto. Dopo la nascita i livelli plasmatici di DHEAS diminuiscono significativamente e rimangono bassi per i primi cinque anni di vita. I livelli successivamente aumentano e raggiungono il valore più elevato intorno ai 25 anni, per poi diminuire con il progredire dell’età. Intorno ai 70-80 anni si assiste a un calo di circa il 70% rispetto al picco massimo (67).
Si può inoltre notare una differenza nel contenuto dell’ormone a seconda del sesso: nell’uomo sono stati dimostrati livelli plasmatici di DHEAS più elevati rispetto alla donna, mentre i livelli di DHEA sembrano simili tra i due sessi.
A questo steroide sono state attribuite diverse funzioni biologiche, in ambito metabolico, immunitario e cognitivo, e considerata la sua progressiva diminuzione nell’organismo, diversi autori ne hanno proposto l’impiego come supplemento alimentare (68). Numerosi studi epidemiologici passati ed esperimenti condotti sugli animali hanno dimostrato come il DHEA sia un ormone multifunzionale, con proprietà benefiche nella prevenzione del diabete (69,70), dell’obesità (71,72), dell’aterosclerosi (73), dell’osteoporosi e in generale sul prolungamento della vita; inoltre al DHEA è stata attribuita capacità di modulare il sistema immunitario (74) e il sistema nervoso centrale (75) e non ultima, di rilevante interesse, un’azione di anticarcinogenesi (76,77).
DHEA e tumore
Bassi livelli plasmatici di DHEA sono significativamente correlati ad un aumentato rischio di insorgenza di tumore con il progredire dell’età (78). Il DHEA possiede capacità anti-proliferative e chemiopreventive nei confronti di diversi tumori, come dimostrato in numerosi lavori. Lo steroide è in grado di rallentare la carcinogenesi indotta chimicamente in differenti tessuti, tra cui il fegato (79), il colon (80), il polmone (81) e la ghiandola mammaria (82), e inibisce la proliferazione di cellule tumorali (83), di cellule non tumorali (84) e di linee cellulari derivate da tumori ( 85).
DHEA e sistema nervoso centrale
Il DHEA e il DHEAS sono stati classificati all’interno del gruppo degli steroidi definiti come neurosteroidi, così chiamati dal momento che possono essere sintetizzati de novo nel sistema nervoso centrale in maniera indipendente (fig 7), almeno in parte, dall’attività degli organi periferici (86); la concentrazione di questi steroidi è molto più alta nel cervello rispetto agli altri organi (87).
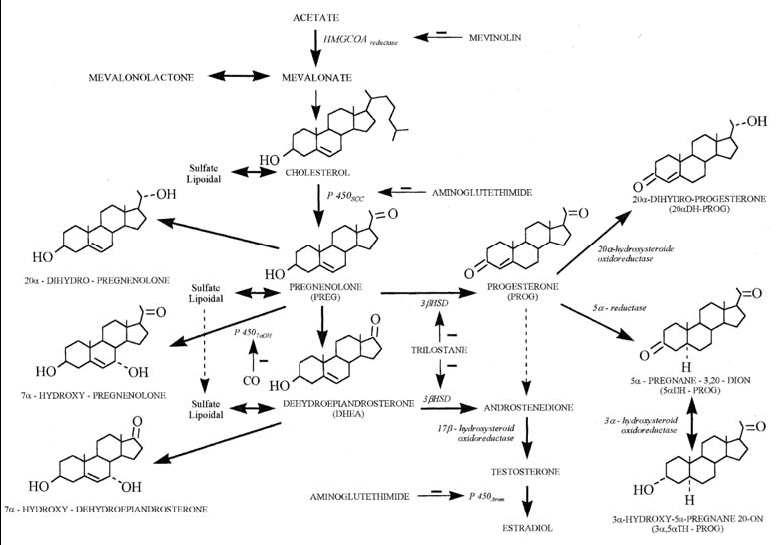
Fig 7. Biosintesi dei neurosteroidi
I tentativi di stabilire la presenza di una associazione tra livelli plasmatici di DHEAS e malattie degenerative del sistema nervoso centrale, tra cui demenza e diminuzione delle capacità cognitive, hanno portato fino ad ora a risultati controversi (88). Le indagini condotte sul potenziale ruolo di questi ormoni nel sistema nervoso centrale hanno evidenziato una loro azione modulatoria a carico di due importanti recettori: il recettore GABA-R (γ-aminobutirric acid receptor) a cui si lega l’acido γ-amminobutirrico, principale neurotrasmettitore inibitorio e il recettore NMDA, a cui si lega N-metil-D-aspartato, fondamentale neurotrasmettitore eccitatorio (89).
Numerosi studi in vitro hanno evidenziato una funzione neuroprotettiva esercitata dallo steroide: il DHEAS previene la morte neuronale per citotossicità indotta in seguito all’aggiunta di glutammato nel mezzo di coltura (90); protegge le cellule ippocampali dal danno cellulare causato da stress ossidativo (91); stimola specifiche vie di trasduzione del segnale coinvolte nei processi di sopravvivenza cellulare (90,92,93,94).
Meccanismi d’azione del DHEA
Il ruolo fisiologico del DHEA non è stato ancora del tutto chiarito, sebbene sia il più abbondante steroide plasmatico.
Il DHEA esercita un’azione indiretta, in seguito alla sua conversione in androgeni ed estrogeni, sui tessuti periferici bersaglio degli ormoni sessuali steroidei, ed un’azione diretta come steroide. In entrambi i casi, il meccanismo può essere di tipo genomico, attraverso la via classica di attivazione dei recettori per gli ormoni steroidei, o di tipo non genomico (95).
Per quanto riguarda il meccanismo d’azione genomico, non sono stati ancora isolati recettori nucleari o recettori di altro genere specifici per il DHEA o per il DHEAS; sono stati descritti siti di legame ad alta affinità per il DHEA a livello dei linfociti T umani e murini (96), ma questi siti non sono specifici e legano anche il diidrotestosterone. Alcuni autori hanno avanzato l’ipotesi di un’interazione tra DHEA e recettore per gli estrogeni (97) (figura 8).
Una risposta genomica è stata osservata nei roditori in cui il DHEA agisce da proliferatore dei perossisomi inducendo l’espressione di geni di enzimi microsomiali e perossisomali attraverso l’attivazione del recettore nucleare PPARα (peroxisome proliferator-activated receptor α) (102). In questo modo il DHEA potrebbe servire come regolatore endogeno del segnale mediato da PPARα a livello epatico, mantenendo l’omeostasi lipidica e prevenendo il declino dell’espressione di PPARα durante l’invecchiamento. Recenti lavori hanno comunque dimostrato che il DHEA, indipendentemente dall’interazione con il recettore PPARα, può modulare l’espressione di diversi geni tra cui quello per il citocromo P450 3A23, enzima coinvolto nel metabolismo di farmaci e sostanze xenobiotiche (103).
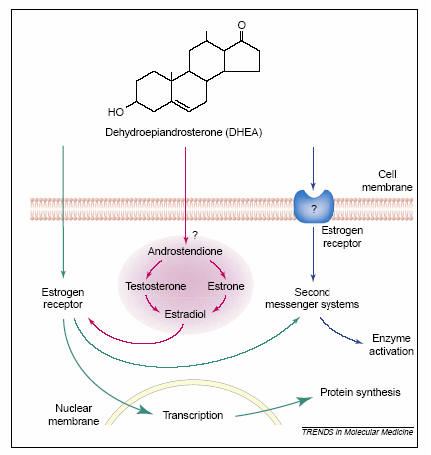
Fig 8. Possibili meccanismi d’azione del DHEA coinvolgenti il recettore per gli estrogeni.
Il DHEA potrebbe legarsi ai recettori degli estrogeni citosolici: in seguito ad attivazione il recettore attraversa la membrana nucleare e induce la sintesi ex novo di proteine specifiche e/o attiva secondi messaggeri che modulano l’attività di specifici enzimi (ruota verde). Dopo aver attraversato la membrana plasmatica, il DHEA potrebbe essere convertito all’interno della cellula in estradiolo, che lega il recettore degli estrogeni inducendo gli effetti sopra citati (ruota rossa). Il DHEA potrebbe legarsi a specifici siti di legame sulla membrana plasmatica (ad es. recettore degli estrogeni), modulando l’attività di enzimi attraverso un sistema di secondi messaggeri (ruota blu) .
Studi condotti su cellule endoteliali umane (HUVEC) hanno dimostrato che il DHEA è in grado di modulare l’espressione dell’enzima monossido di azoto sintasi endoteliale (eNOS), inducendone una maggiore espressione attraverso un meccanismo diretto genomico; l’ormone inoltre stabilizza la proteina riducendone il suo turnover. Oltre all’effetto genomico diretto, il DHEA nelle HUVEC sembra esercitare anche un effetto non genomico, portando a un significativo aumento dell’attività enzimatica della eNOS, valutabile pochi minuti dopo lo stimolo con l’ormone (104).
Altri meccanismi d’azione di tipo non genomico sono stati osservati nel sistema nervoso. Il DHEA è in grado di interagire con i recettori di alcuni neurotrasmettitori, funzionando da antagonista del recettore dell’acido gamma aminobutirrico (GABA-R): esperimenti condotti su colture cellulari di neuroni hanno dimostrato che in vitro il DHEA si comporta da modulatore non competitivo del GABA-R, inibendo il trasporto di ioni cloro in maniera dose dipendente, con conseguente riduzione dell’iperpolarizzazione di membrana e dell’eccitabilità neuronale. Lo steroide sembrerebbe inoltre potenziare l’azione di N-metil-D-aspartato (NMDA), comportandosi da agonista della subunità sigma-1 del recettore. In generale il DHEA è considerato un neurosteroide eccitatorio naturale (44).
Lavori molto interessanti hanno attribuito ai neurosteroidi un ruolo nella formazione del citoscheletro cellulare e in particolare dei microtubuli. E’ stato osservato che il pregnenolone (PREG), il neurosteroide più abbondante, è in grado di associarsi alle MAP (microtubule-associated protein). I prolungamenti neuritici dei neuroni contengono un elevato numero di microtubuli paralleli al loro interno. Questi giocano un ruolo importante nella crescita e nel mantenimento dei neuriti durante il differenziamento neuronale. Le MAP costituiscono i microtubuli insieme con la tubulina e rappresentano i maggiori componenti citoplasmatici nel neurone. Determinano la forma neuronale e il bilancio tra rigidità e plasticità nei processi neuronali. PREG sembra essere in grado di aumentare l’efficienza dell’assemblaggio dei microtubuli grazie al legame con MAP-2 (marker dendritico). Sono riportati comportamenti analoghi per gli altri neurosteroidi. Il DHEAS incrementa selettivamente la lunghezza dei dendriti contenenti MAP-2, mentre il DHEA incrementa la lunghezza dei neuriti contenenti il marker assonale Tau (anch’esso appartenente alla famiglia delle MAP). Non è ancora chiaro se DHEA e DHEAS agiscano con lo stesso meccanismo di PREG (44).
I meccanismi molecolari attraverso cui il DHEA esercita un’azione anti-proliferativa non sono ancora perfettamente chiariti: alcuni autori ritengono che lo steroide potrebbe comportarsi da potente inibitore non competitivo della glucosio-6-fosfato deidrogenasi (G6PD), enzima limitante il ciclo dei pentoso fosfati. Il blocco di questa via metabolica porterebbe a una diminuzione di riboso fosfato e della quantità di NADPH all’interno della cellula, necessari per la sintesi di ribonucleotidi e desossiribonucleotidi per la replicazione cellulare (98).
Un altro caso in cui il DHEA agirebbe con meccanismo non genomico è l’inibizione della via di biosintesi del colesterolo e la via di trasduzione del segnale mediato dalle proteine cinasi attivate da mitogeni (MAPK, mitogen-activated protein kinase), attraverso l’inibizione dell’attività dell’enzima 3-idrossimetilglutaril-coenzimaA reduttasi (HMG-CoA reduttasi) (99). HMG-CoA reduttasi è l’enzima limitante la biosintesi del farnesil difosfato, derivato isoprenoide precursore del colesterolo e indispensabile per la farnesilazione della proteina Ras.
Nel segnale di proliferazione cellulare mediato dalle MAPK interviene Ras, che una volta attivata (farnesilata e legata al GTP) è in grado a sua volta di attivare la cinasi Raf1 che fosforila la cinasi MEK1, rendendola attiva e in grado di fosforilare ERK1 e ERK2. Queste MAPK possono fosforilare una grande varietà di substrati tra cui le proteine Fos e Jun, (rispettivamente i prodotti dei proto-oncogeni c-fos e c-jun), che risultano essere i componenti del fattore trascrizionale AP-1 (activator protein-1). Il legame di AP-1 a siti regolatori sul DNA determina l’attivazione trascrizionale di numerosi geni che mediano la proliferazione e la progressione nel ciclo cellulare attraverso l’inizio di sintesi di nuovo DNA (100). A questo proposito, studi recenti hanno dimostrato che in colture di cellule muscolari lisce delle vie aeree, il trattamento con DHEA causa una significativa diminuzione del legame di AP-1 al DNA; probabilmente l’effetto sembra essere causato dall’inibizione di una proteina cinasi che media la fosforilazione di AP-1, evento necessario affinchè il fattore trascrizionale si possa legare al DNA (100).
Ad ogni modo, l’effetto anti-proliferativo del DHEA, spiegato attraverso l’inibizione degli enzimi G6PD e HMG-CoA reduttasi, tuttora non è considerato dalla letteratura del tutto convincente, al punto da suggerire un possibile coinvolgimento di altri meccanismi molecolari ancora da indagare (101).
Le Metalloproteasi
La formazione di neuriti, marker del differenziamento neuronale, implica una riorganizzazione del citoscheletro e un’invasione della matrice extracellulare (ECM). Le metalloproteasi (MMP) sono componenti essenziali del sistema enzimatico responsabile della degradazione dell’ECM, sistema che consente la migrazione neuronale e la crescita dei neuriti durante i processi di normale riparazione dei nervi o di invasione tumorale (28,29).
Le MMP sono una famiglia di enzimi con più di 20 membri identificati, tutti caratterizzati da un’omologia di sequenza e da una similarità strutturale ed enzimatica. Sono endopeptidasi extracellulari che richiedono Zn2+ per la loro attività. I loro maggiori targets sono, appunto, le proteine della matrice extracellulare. Le MMP diventano attive in seguito a taglio del corrispondente propeptide e, una volta attive, possono essere inibite dal legame non covalente con degli inibitori specifici (TIMP) (105).
La regolazione dell’espressione e dell’attività delle MMP è un processo molto complesso. Il caso della MMP-9 è rappresentato in fig 9.
Il gene di MMP-9 viene trascritto in seguito a vari attivatori (tra cui AP-1) e la stabilità dell’mRNA viene regolata da NO. La proteina, inizialmente sintetizzata come pre-pro-MMP-9, migra nel reticolo endoplasmatico in cui diventa pro-MMP-9 e subisce modificazioni post-traslazionali (glicosilazioni) che la regolano ulteriormente. A questo punto viene secreta e, una volta giunta nello spazio extracellulare, viene definitivamente tagliata con attivazione del sito catalitico contenente Zn. L’attivazione del pro-enzima è controllata da una cascata di passaggi che coinvolge anche altre metalloproteasi. Si noti come MMP-2 sia in grado di attivare MMP-9 (105)

Fig 9. Espressione e attivazione di MMP-9
La MMP-2 (o gelatinasi A) di 72 kDa e la MMP-9 (o gelatinasi B) di 92 kDa sono collagenasi di tipo IV di particolare importanza nei processi di invasione cellulare (30,31).
L’effetto differenziante dell’acido retinoico include anche il controllo dei geni delle MMP: RA promuove l’espressione sia di MMP-2 che di MMP-9 (32-34). Inoltre esistono lavori che dimostrano come anche il DHEA sia in grado di stimolare la produzione delle MMP (35).
Le Transglutaminasi
Come le metalloproteasi, anche le transglutaminasi (TG) sono una classe di proteine il cui ruolo risulta fondamentale nei processi di migrazione e di allungamento dei neuriti (56).
Le TG vengono distinte in base alle loro proprietà fisiche, alla distribuzione e localizzazione nei tessuti, ai substrati e ai meccanismi di attivazione e di azione. Nonostante la presenza nella loro struttura di siti di glicosilazione e di residui cisteinici, non subiscono glicosilazioni e non presentano ponti disolfuro (106).
Tutte le transglutaminasi richiedono un elevata concentrazione di Ca2+ per l’attivazione. Tra le reazioni catalizzate dalle TG, la più studiata e nota è il crosslinking tra proteine. Molto significative sono anche altre modificazioni post-traduzionali mediate da TG, come la deaminazione e l’incorporazione di ammine (106). Un elenco che riassume le principali reazioni catalizzate da TG si trova in fig 10.
La più importante di questa famiglia di enzimi è la TG2. Nonostante sia stata la prima ad essere scoperta, rimangono ancora molti dubbi sul suo ruolo fisiologico. Si tratta di una proteina prevalentemente citosolica (80%), localizzata in misura minore nelle membrane nucleari (5%); nell’organismo è contenuta anche nel plasma (10-15%). Viene secreta dalla cellula con un meccanismo ancora sconosciuto, per distribuirsi sulla superficie cellulare e nell’ECM.
Una sua funzione molto importante consiste nel mediare l’interazione tra le integrine della membrana cellulare e la fibronettina della matrice extracellulare, attraverso un’associazione diretta con le integrine (β1 e β3 ) e con la fibronettina (56) (vedi fig 11).
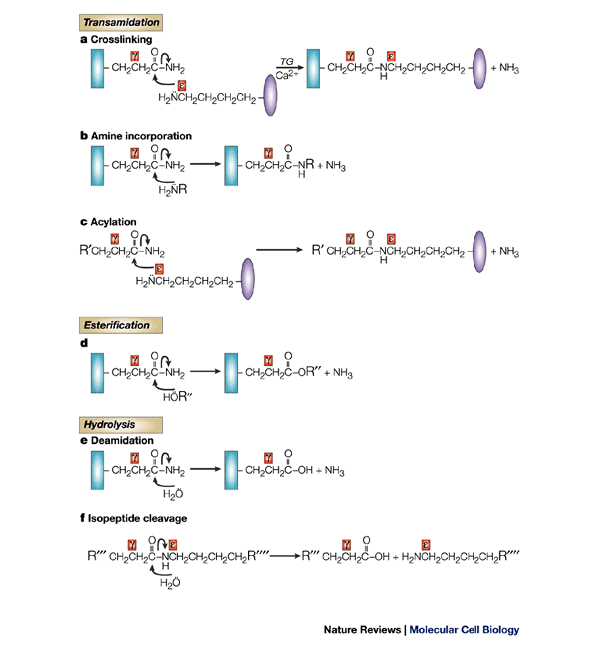
Fig 10. Modificazioni post-traduzionali catalizzate da transglutaminasi
Con questa funzione le TG regolano l’adesione e lo spreading cellulare, quindi ricoprono una notevole importanza in molti fenomeni tra cui la produzione dei neuriti nei neuroni. Esistono lavori, ancora in fase di discussione, che correlano la presenza delle TG con la patogenesi di malattie neurodegenerative. Sembra invece evidente che una ridotta espressione di transglutaminasi sia associata ad una maggiore aggressività tumorale (106). Le TG vengono indotte da acido retinoico con il coinvolgimento dei recettori RARα e RARγ (57,58). E’ stata dimostrata una implicazione di questi recettori nella crescita dei prolungamenti neuritici in cellule di neuroblastoma (59). Inoltre è fondamentale la loro interazione con le MMP, il cui ruolo proteasico nei confronti delle TG di membrana regola l’adesione e il movimento delle cellule tumorali (60).
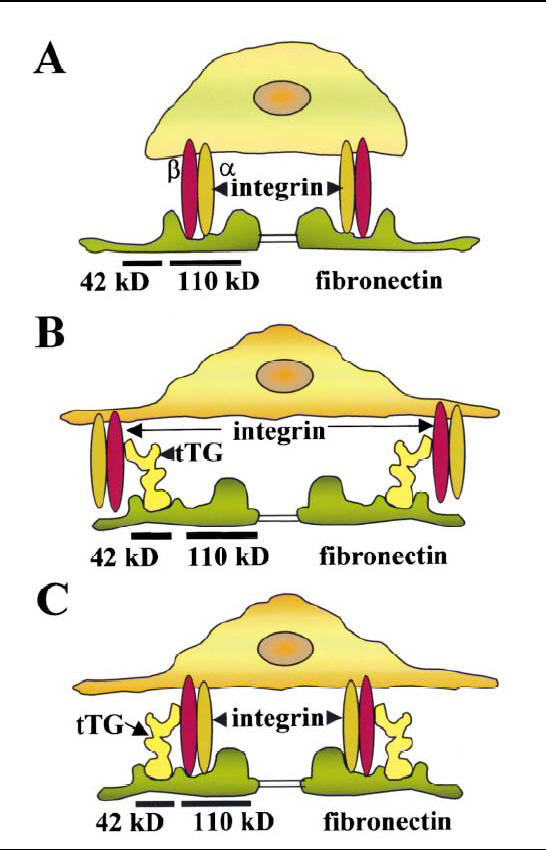
Fig 11. Ruolo delle transglutaminasi nell’adesione cellulare
(A) Adesione alla fibronettina (Fn) attraverso le integrine in assenza di TG. (B) Le TG facilitano l’adesione funzionando da ponte tra integrine e Fn. (C) Formazione di un complesso in cui le tre proteine interagiscono una con l’altra.
Scopo del lavoro
L’acido retinoico è un noto agente differenziante del tessuto nervoso. Molti dei suoi meccanismi d’azione sono noti. Durante lo sviluppo è particolarmente importante l’azione di RA in sinergia con altre molecole segnale, tra le quali i neurosteroidi, il cui capostipite, il DHEA, è prodotto in grande abbondanza ed esercita funzioni neuroprotettive e differenzianti.
Questo studio ha voluto analizzare una possibile cooperazione tra RA e DHEA nel programmare le cellule nervose verso il differenziamento. Abbiamo utilizzato una linea di neuroblastoma umano (SK-N-BE) in grado di differenziare in seguito al trattamento con acido retinoico. E’ stato quindi condotto uno studio sull’effetto di RA e DHEA sul differenziamento delle cellule di neuroblastoma, in termini di proliferazione e movimento cellulare, formazione di neuriti e di markers neuronali (GAP-43). Per potere poi interpretare i dati ottenuti sono stati presi in considerazione la modulazione dell’espressione di recettori nucleari e di proteine coinvolte nella formazione e allungamento dei neuriti (metalloproteasi e transglutaminasi).
Capitolo 2
PROCEDURE SPERIMENTALI
Colture cellulari
Le cellule (SK-N-BE) sono state coltivate utilizzando come mezzo di coltura DMEM (Gibco, Paisley, UK) senza rosso fenolo con 10% FBS, 100000 U/l penicillina, 100 mg/l streptomicina e 2 mM L-glutammina a 37 °C e CO2 al 5%.
Sono state trattate per i tempi riportati con all-trans RA (Sigma Chemical, St. Louis, MO) 10μM, DHEA (Fluka, Buchs, Switzerland) 100 μM, RA e DHEA, con periodica sostituzione del mezzo.
Proliferazione cellulare
Le cellule sono state seminate in una piastra da 24 pozzetti in presenza delle due sostanze da sole o insieme. Dopo 9 giorni di incubazione, le cellule sono state colorate con crystal violet 0.1% in metanolo 20% per 20 min, lavate con acqua e quindi solubilizzate con acido acetico 10%. L’assorbanza è stata misurata a 590 μm utilizzando un fotometro microplate (EL-340 Bio-Tek Instrument, Winooski, VT, USA).
Movimento e crescita dei neuriti
Dopo 3 giorni di trattamento in fiasche T-25 il terreno é stato rimpiazzato da un mezzo contenente Hepes (SGG medium: 114 mM NaCl, 26.1 mM NaHCO3, 5.3 mM KCl, 1 mM MgCl2, 2 mM CaCl2, 10 mM Hepes, 1 mM glycine, 30 mM glucose, 0.5 mM sodium pyruvate, 0.001% phenol red). Il movimento cellulare è stato studiato per 12 ore utilizzando un microscopio Nikon Diaphot con obiettivo a contrasto di fase 10X, mantenedo le cellule in un incubatore
(Plexiglas Nikon NP-2) a 37 °C. La migrazione è stata registrata da una videocamera JVC-1CCD. Le immagini acquisite sono state analizzate con il sistema Microimage (Cast Imaging, Venice, Italy) in modo da ricostruire il movimento che è stato quindi quantificato moltiplicando la lunghezza del percorso compiuto dalla cellula con la distanza tra la posizione iniziale e quella finale. Inoltre, il primo e l’ultimo frame sono stati confrontati per misurare la crescita dei neuriti nel corso delle 12 ore.
Immunofluorescenza
Le cellule sono state coltivate su vetrini trattati con poli-lisina. Al termine dei trattamenti (9 giorni), sono state fissate in paraformaldeide 3%, saccarosio 4% in PBS a temperatura ambiente per 20 min. Dopo permeabilizzazione con NP40 0.5% in PBS per 5 min, le cellule sono state bloccate in normal goat serum 1.5% in PBS per 20 min e successivamente trattate overnight con un anticorpo policlonale anti-GAP43 (dil. 1:400; Chemicon International, Temecula, CA, USA). Dopo i lavaggi, sono state incubate con anticorpo secondario biotinilato e con fluorescin-avidin D. Le cellule sono state osservate con un microscopio Olympus a fluorescenza e fotografate con un sistema digitale Olympus DP10.
Western Blot
Le cellule trattate sono state raccolte e lisate in sample buffer 1X (100 mM Tris-HCl pH 6.8, 0.01% bromophenol blue, 2% SDS, 15% glycerol, 10 mM DTT). Nella frazione solubile, addizionata degli inibitori delle proteasi, é stata valutata la concentrazione di proteine totali. Uguali quantità di proteine (30 μg) sono state caricate e separate in un gel di poliacrilamide al 10%. Dopo trasferimento su membrana PVD, sono state bloccate, incubate con l’anticorpo primario (tutti gli anticorpi da Santa Cruz Biotechnology, CA, USA) per 1 h a temperatura ambiente e, successivamente, con il secondario coniugato con perossidasi. Le proteine di interesse sono state evidenziate tramite ECL (Perkin Elmer Life Science, Boston, MA, USA).
Analisi zimografica
L’analisi è stata effettuata sia sul terreno di coltura posto a contatto con le cellule sia sugli estratti cellulari. Le cellule sono state lisate nel tampone di lisi (1% Triton X-100, 50 mM Tris, 300 mM NaCl, pH 7.5). Le proteine contenute nei campioni sono state separate per elettroforesi in un gel di poliacrilamide all’8% contenente gelatina (0.1%) in condizioni non riducenti. Il gel è stato successivamente lavato con Triton X-100 2.5% per rimuovere l’SDS e incubato a 37 °C in collagenase buffer (50 mM Tris-HCl, 200 mM NaCl, 10 mM CaCl2, pH 7.5) per 16h o 5 giorni. Al termine i gel sono stati colorati con Coomassie Brillant Blue-250 e decolorati in acqua.
Biotinilazione della superficie cellulare
Al termine dei trattamenti le cellule sono state lavate con KRPB (128 mM NaCl, 4.7 mM KCl, 1.25 mM CaCl2, 1.25 mM MgSO4, 5 mM NaH2PO4) contenente 20mM Hepes, pH 7.4. Sono state poi trattate con sulfo-NHS-LC-biotin (Pierce) 1mM in agitazione a 4°C per 15’. Successivamente sono state lavate con un tampone di lavaggio (25 mM etanolammina, 150 mM NaCl, 20 mM Tris-Cl, pH 7.4) e lisate nel tampone di lisi (1% octaethylene glicol dodecyl ether, 150 mM NaCl, 20 mM Tris-Cl, pH 7.4) in presenza di inibitori di proteasi. E’ stata determinata la concentrazione delle proteine totali nei lisati e aliquote sono state immunoprecipitate a 4°C con l’aggiunta dell’anticorpo anti-TG (NeoMarkers) per 12h. L’immunoprecipitato è stato raccolto con Protein A/G Plus-agarose (Santa. Cruz) a 4°C per 2h e solubilizzato in tampone contenente SDS. I campioni sono stati infine separati tramite western blotting. Le proteine biotinilate sono state visualizzate con ECL dopo trattamento con streptavidin-HRP.
Analisi statistica
I dati sono stati espressi come media ± SEM. L’analisi statistica è stata condotta utilizzando il test t di Student.
Capitolo 4
RISULTATI
RA e DHEA inibiscono la proliferazione cellulare
Sia RA che DHEA sono noti mediatori dell’arresto proliferativo. Abbiamo quindi verificato il loro effetto sulle SK-N-BE. Attraverso la tecnica di colorazione con crystal violet è stata valutata la crescita delle cellule non trattate e di quelle trattate con all-trans RA (10 μM), con DHEA (100 μM) e con una combinazione delle due sostanze. In fig. 12 si può osservare che i nostri dati confermano un effetto antiproliferativo di RA e DHEA sulle cellule di neuroblastoma; questo effetto si manifesta in misura ancora più evidente quando le due sostanze vengono utilizzate insieme.
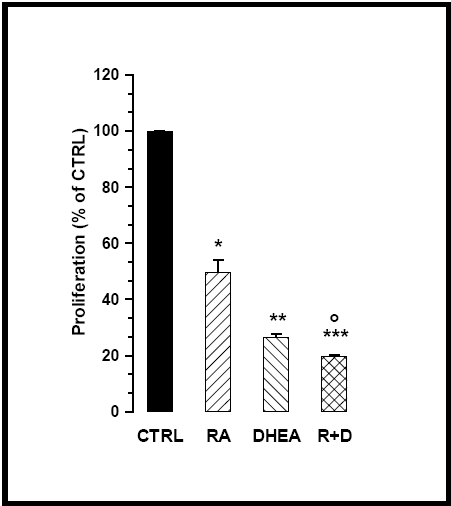
Fig. 12 – Proliferazione cellulare
I valori sono espressi come percentuale del controllo
(* p<0.01 vs ctrl, ** p<0.001 vs ctrl, *** p<0.0001 vs ctrl,° p<0.05 vs RA e DHEA)
Effetto di RA e DHEA sulla motilità cellulare
Le cellule sono state osservate per 12 h come descritto in Materiali e Metodi. I percorsi compiuti sono visibili in fig.13 in cui si può osservare come le cellule del controllo abbiano un movimento direzionale mentre quelle trattate assumono un moto stazionario intorno ad un punto fisso. Inoltre, come è stato quantificato in fig.14, RA e DHEA riducono la motilità cellulare.
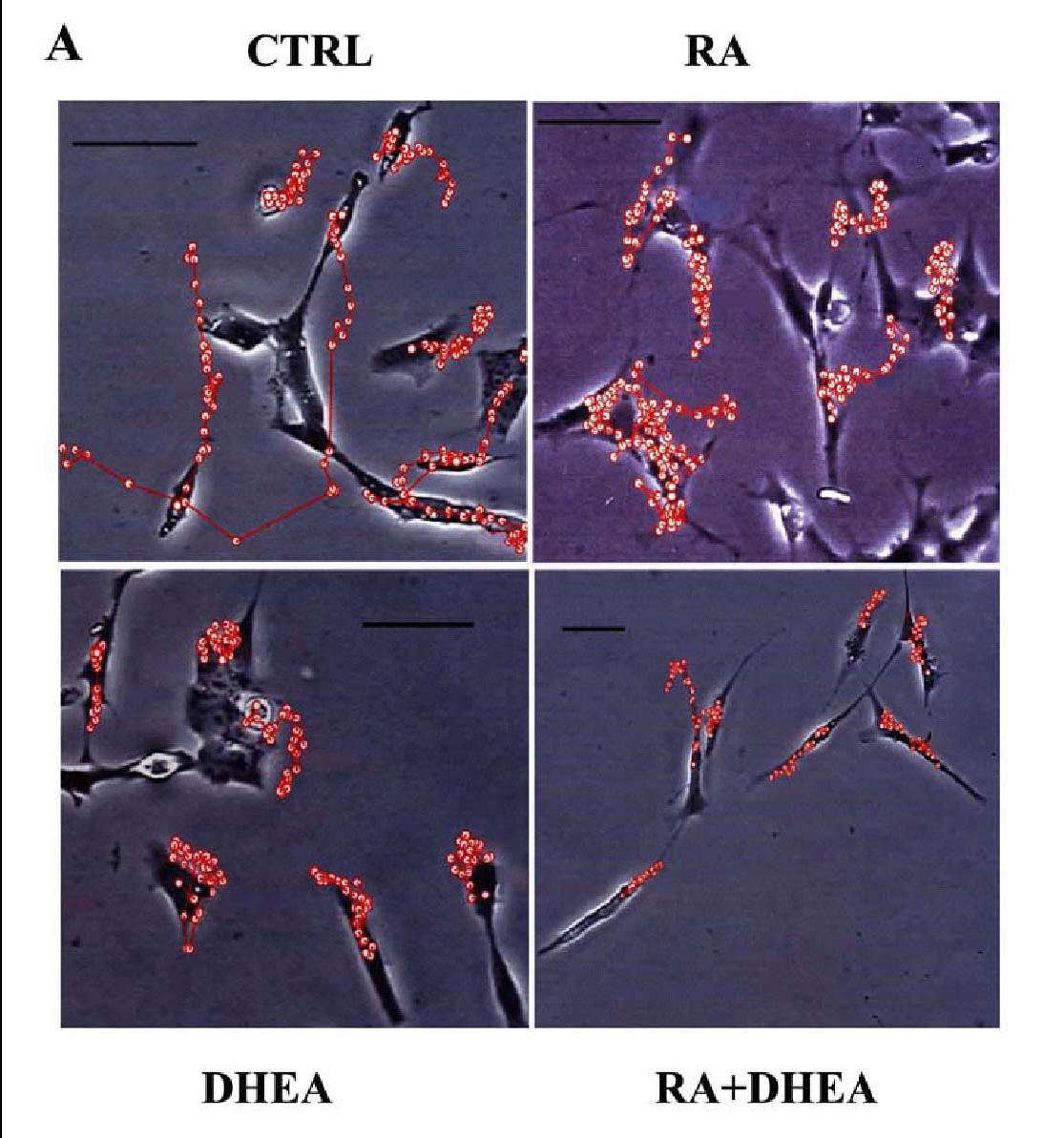
Fig 13. Movimento cellulare
In rosso sono rappresentati i percorsi compiuti dalle SK-N-BE nel corso delle 12 h di osservazione. Le fotografie sono rappresentative di un totale di tre esperimenti (Bar=100 μm)
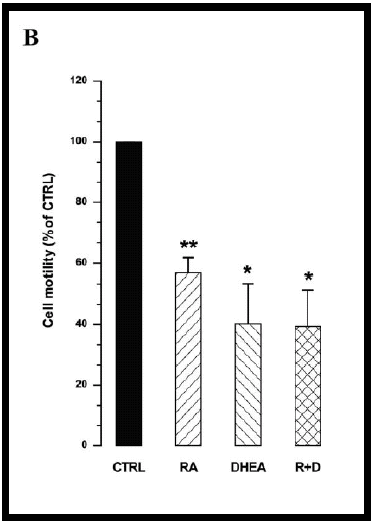
Fig. 14– Motilita’ cellulare
Il valore della motilità cellulare è stato ottenuto moltiplicando la lunghezza del percorso con la distanza tra il punto iniziale e finale. Nel grafico i valori sono espressi come percentuale del controllo nella forma Media ± S.E.M. (n=3)
(* p<0.02 vs ctrl, ** p<0.005 vs ctrl)
RA e DHEA sinergizzano nel promuovere il differenziamento cellulare
Il differenziamento è stato valutato in termini di formazione dei neuriti e di espressione della proteina marker neuronale GAP-43 (growth associated protein-43).
I neuriti sono stati osservati al 3° giorno di trattamento e al termine delle 12 ore successive. La fig. 15A mostra degli esempi di cellule fotografate al tempo 0 e dopo 12 h. Dal grafico che rappresenta la lunghezza iniziale dei neuriti (fig. 15B) è evidente l’effetto di RA sul differenziamento, mentre dal grafico che mostra l’allungamento nel corso delle 12 h (fig. 15C) risulta evidente l’effetto sinergico di RA e DHEA.
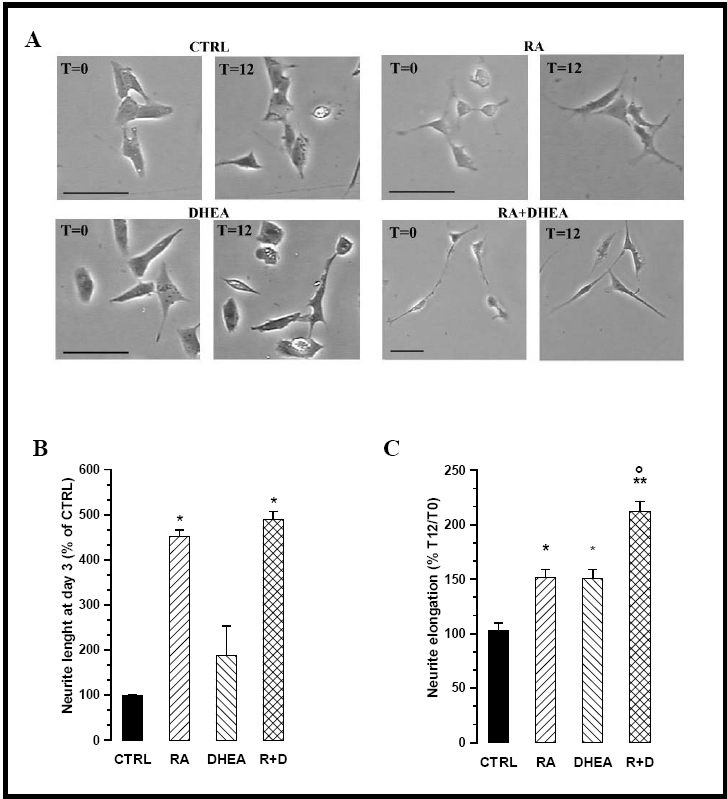
Fig 15 – Produzione di neuriti promossa da RA e DHEA
A. Le fotografie mostrano un campo raffigurante le cellule di controllo e quelle trattate, sia al tempo 0 (T=0) che dopo 12 h (T=12).
B. Nel grafico sono riportate le lunghezze misurate dei neuriti al T=0 (corrispondente al 3° giorno di trattamento) espresse come % del CTRL.
C. L’allungamento dei neuriti nel corso delle 12 h è stato calcolato facendo il rapporto tra la loro lunghezza al T=0 e al T=12
(I valori sono indicati come Media ± S.E.M. per n=3. * p<0.02 vs ctrl, ** p<0.002 vs ctrl, ° p<0.01 vs RA e DHEA. Bar = 100 μm)
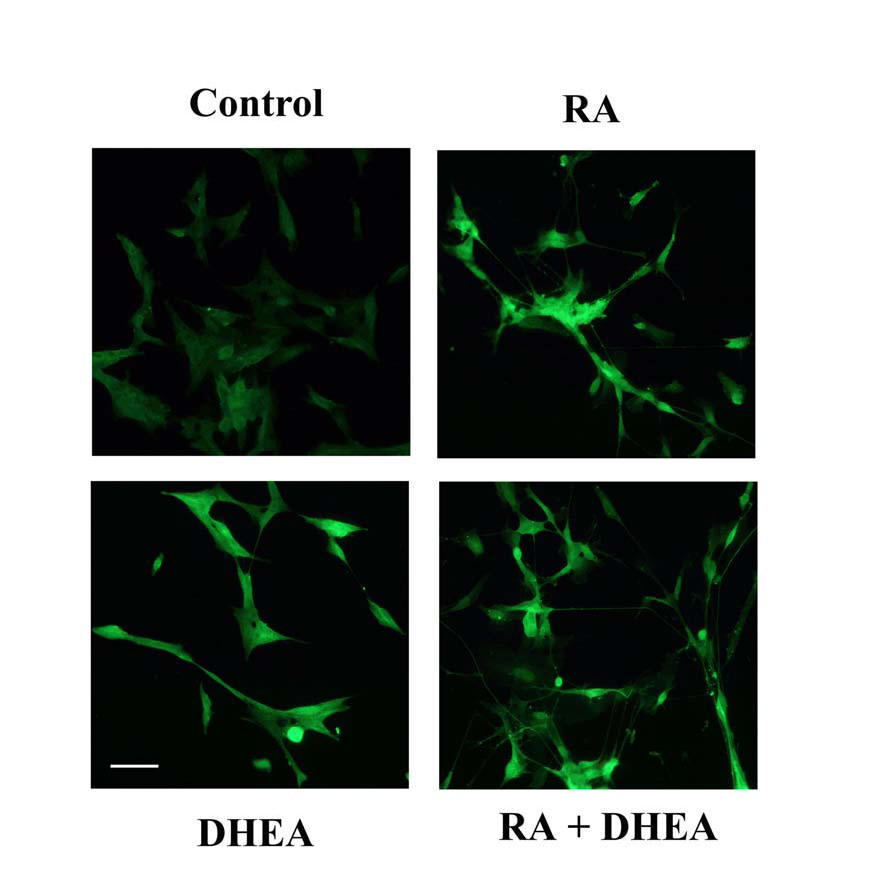
L’esperimento di immunofluorescenza mostrato in fig.16 permette di osservare una maggiore espressione della GAP-43 nelle cellule trattate contemporaneamente con RA e DHEA, tra le quali si può osservare dalla fotografia l’elevato numero di connessioni neuritiche.
Fig. 16 –Espressione di GAP-43 attraverso immunofluorescenza
Le cellule sono state trattate su vetrini per 9 giorni con RA, DHEA e RA+DHEA. Successivamente sono state incubate con anticorpo anti-GAP43. Le immagini riportate in fig sono rappresentative di 3 esperimenti (Bar=100 μm)
L’azione sinergica di RA e DHEA sul differenziamento del neuroblastoma non è mediata da un’alterazione dell’espressione dei recettori per l’acido retinoico
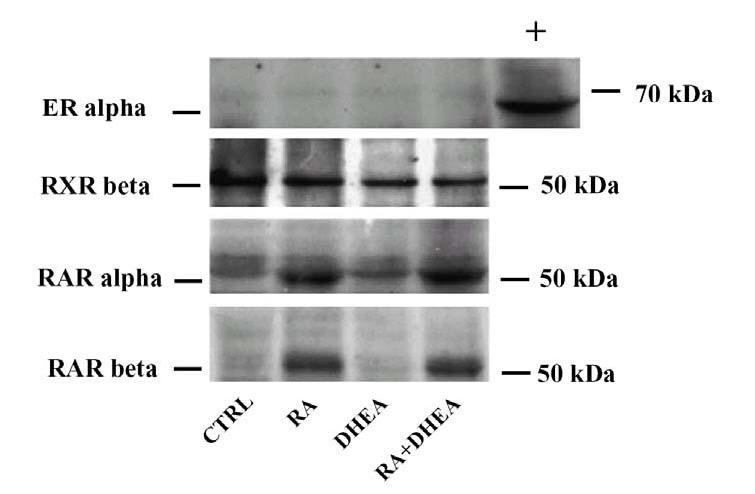
Per poter spiegare l’effetto sinergico di RA e DHEA sul differenziamento è stato valutato il coinvolgimento dei recettori nucleari responsabili del processo di differenziamento. A tale scopo è stata valutata l’espressione di due recettori della famiglia dei RAR, le isoforme α e β, noti per essere mediatori dell’effetto apoptotico e antiproliferativo dei retinoidi, e uno dei recettori che modula la formazione di eterodimeri, RXRβ. Inoltre si è controllata la presenza nelle nostre cellule del recettore per gli estrogeni ERα, uno dei possibili recettori per i metaboliti del DHEA. In fig.17 è mostrata l’analisi western blot dell’espressione dei recettori dopo 9 giorni di trattamento: sia l’espressione di RARα che di RARβ è incrementata dall’acido retinoico, mentre non è influenzata dalla presenza del DHEA, sia da solo che in combinazione con RA. L’espressione di RXRβ non sembra essere modulata da nessuna delle due sostanze. ERα non è presente in quantità rivelabile nelle SK-N-BE per cui è da escludere un suo coinvolgimento nell’azione del DHEA.
Modulazione dell’attività metalloproteasica durante il differenziamento
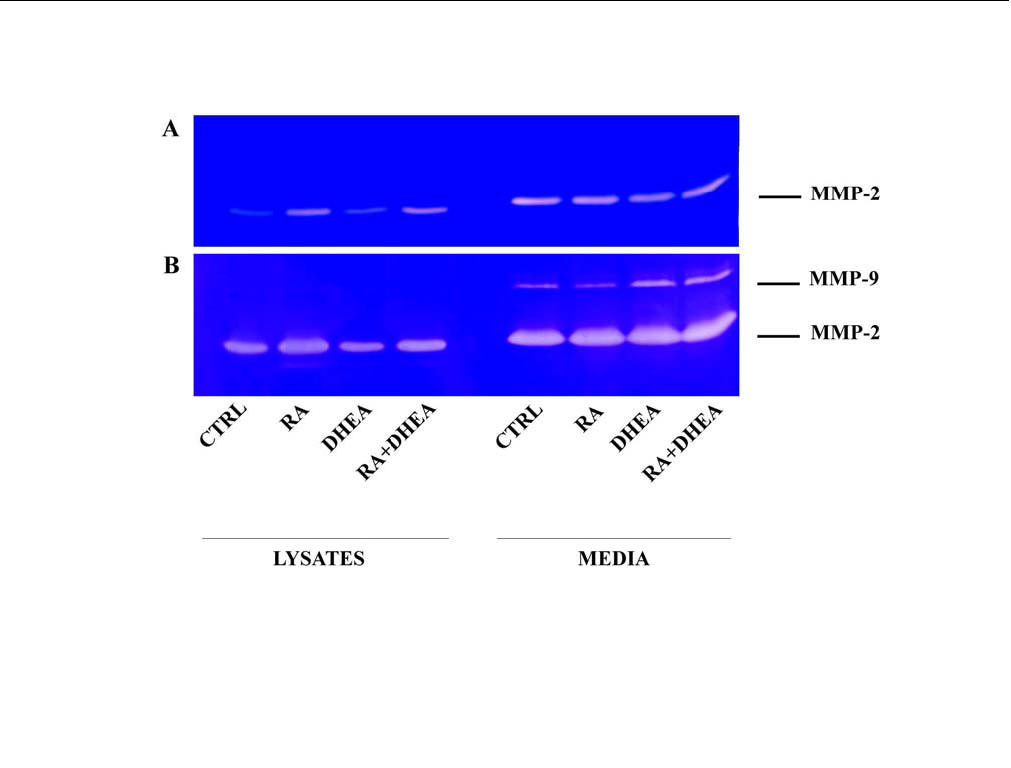
La produzione delle metalloproteasi, legate alle membrane o secrete dalla cellula, è uno dei meccanismi implicati nell’invasione della matrice extracellulare e nella migrazione cellulare. Abbiamo quindi valutato l’attività delle metalloproteasi, sia legate alle cellule che secrete dalle cellule, prodotte in seguito ai trattamenti con RA e DHEA. La MMP-2 è determinabile zimograficamente sia nei lisati cellulari che nel terreno condizionato anche dopo breve incubazione (fig.18A). Solo la MMP-2 legata alla cellula mostra un aumento di attività dopo trattamento con RA e RA+DHEA. L’attività della MMP-9 è invece determinabile solo nel terreno extracellulare e dopo prolungata incubazione del gel, indice del fatto che la proteina è totalmente secreta e presente in quantità limitata (fig 18B). Il dato interessante è che l’attività della MMP-2 è incrementata durante il differenziamento promosso dall’acido retinoico, e non dal DHEA, mentre l’attività della MMP-9 è aumentata del DHEA e non dal RA. Il sinergismo tra queste due sostanze, in particolare per quanto riguarda la produzione dei neuriti e il movimento cellulare, può essere quindi legato ad una simultanea induzione di queste due diverse metalloproteasi.
Fig. 17 – Analisi western blot dei recettori nucleari
Le cellule sono state lisate dopo 9 giorni di trattamento e l’espressione dei recettori (indicati nella parte sinistra) è stata valutata attraverso western blotting. Nel caso di ERα è stato utilizzato anche un estratto di MCF-7 come controllo positivo in modo da confermare sia l’efficienza dell’anticorpo che l’assenza di tale recettore negli estratti di SK-N-BE.
Fig. 18 – Analisi zimografica delle metalloproteasi
Al termine dei trattamenti, l’analisi zimografica è stata condotta sia sul terreno di coltura in cui erano presenti le proteine secrete dalle cellule, sia sui lisati cellulari. La digestione della gelatina è avvenuta per un periodo di 16 h (A) o di 5 giorni (B). Le zone corrispondenti a MMP-2 e MMP-9 sono indicate sulla destra della figura.
Espressione della transglutaminasi
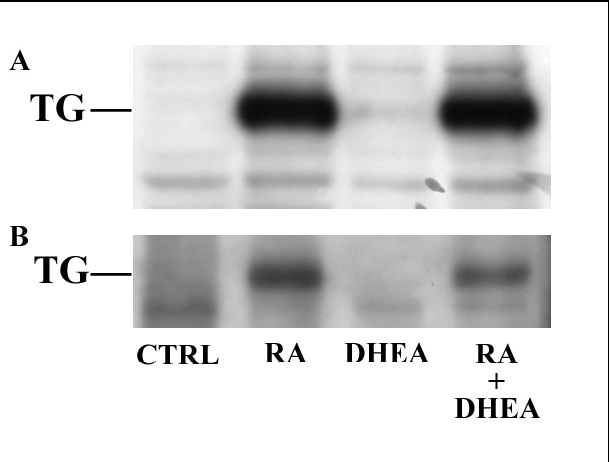
La TG è fondamentale nei processi di adesione e movimento cellulare. Abbiamo valutato l’espressione tramite western blot di questo enzima in cellule trattate. In fig.19A si può notare l’induzione solo da parte di RA, senza alcun effetto del DHEA. Si è quindi ipotizzato che il DHEA pur non modificando l’espressione della TG totale, ne modificasse la traslocazione sulle membrane. Abbiamo quindi voluto focalizzare la nostra attenzione sulle TG di membrana. Le cellule trattate sono state incubate con biotina allo scopo di marcare solo le proteine di superficie, da cui è stata isolata la TG tramite immunoprecipitazione. In fig. 19B è possibile notare che anche sulle TG di membrana si ha solo un effetto di RA, che il DHEA non sembra influenzare.
Fig. 19 – Espressione della transglutamminasi
Le TG totali presenti nelle cellule in seguito ai trattamenti sono state evidenziate tramite western blotting (A). L’entità della loro localizzazione sulle membrane cellulari è stata valutata attraverso biotinilazione (B).
Capitolo 4
DISCUSSIONE
Il differenziamento è un processo che in molti casi le cellule tumorali hanno perso. Il neuroblastoma deriva da neuroblasti simpatici primitivi nella midollare surrenale o nei gangli simpatici ed è caratterizzato da una grande eterogeneità. La possibilità di studiare il differenziamento in vitro in seguito al trattamento con varie molecole ha fatto sì che le linee cellulari di neuroblastoma siano molto utilizzate per indagare sui meccanismi molecolari coinvolti nel differenziamento. L’acido all-trans retinoico è particolarmente efficace nell’indurre l’attivazione del programma di differenziamento, così è stato considerato un promettente agente per la prevenzione e il trattamento di molti tumori non differenziati originati nel sistema nervoso e non solo. Tuttavia molti tipi di tumore manifestano una resistenza ai retinoidi, possono risultare quindi interessanti degli studi su trattamenti combinati.
Considerando ciò, abbiamo voluto cercare eventuali amplificatori dell’effetto dell’acido retinoico sul differenziamento neuronale, trovando che un tale ruolo può essere attribuito ai neurosteroidi. Abbiamo scelto di studiare il DHEA, in quanto viene prodotto dal sistema nervoso e sono note le sue proprietà differenzianti e antitumorali (39,40). Il suo estere solfato DHEAS ha una concentrazione 20 volte maggiore di quella di ogni altro steroide circolante, ma la funzione fisiologica di questo steroide è ancora sconosciuta. In questo studio, dopo trattamento con RA e DHEA, le cellule di SK-N-BE mostrano un rallentamento della proliferazione accompagnato da un aumento dello stato differenziato, osservabile sia come produzione di neuriti che come espressione della proteina marker neuronale GAP-43. Entrambi questi fenomeni, proliferazione e differenziamento, risultano maggiormente modulati dalla co-incubazione con i due farmaci, rispetto all’azione singola di ciascuno di essi.
Inoltre la motilità cellulare diminuisce e diventa più stazionaria. Tutti questi dati concordano nel sostenere un reciproco potenziamento delle proprietà antiproliferative e differenzianti dell’acido retinoico e del DHEA.
Un fenotipo differenziato che manifesta neuriti ben sviluppati deriva dal risultato di cambiamenti della adesione cellulare così come dell’aumento del rimodellamento della matrice extracellulare, fenomeno mediato da proteasi. Anche se cresciute in vitro, le cellule di neuroblastoma sono in grado di secernere varie componenti della matrice come fibronettina, tenascina e laminina (41,42). Nel nostro modello sperimentale l’incrementato allungamento dei neuriti dopo coincubazione con RA e DHEA è accompagnato da una modulazione dell’attività delle proteasi. Infatti il trattamento con RA incrementa l’attività della metalloproteasi MMP-2, mentre il DHEA incrementa quella della MMP-9. Questo dato suggerisce che l’effetto manifestato dal DHEA di potenziamento delle proprietà differenzianti di RA, almeno per quanto riguarda la formazione dei neuriti, è legato ad un più efficiente rimodellamento della matrice extracellulare. La cooperazione tra le due sostanze sembra invece non dipendere da un effetto sull’espressione della tranglutaminasi. Infatti il DHEA non ne causa modificazioni nell’espressione e nella distribuzione all’interno della cellula. Tale enzima è invece indotto da RA, come noto in letteratura, per partecipare ai meccanismi di adesione legati alla formazione dei neuriti.
Per quanto riguarda il meccanismo molecolare alla base dell’interazione degli effetti di RA e DHEA, abbiamo trovato che apparentemente non è coinvolta una modulazione dell’espressione dei recettori. Infatti i recettori per il RA normalmente considerati negli studi di differenziamento, RARα, RARβ e RXRβ, nei nostri esperimenti mostrano un’induzione da ligando (RARα e RARβ) oppure un’espressione basale (RXRβ) che non viene modificata dal cotrattamento con DHEA. Gli esperimenti condotti non sono in grado di discriminare tra l’azione diretta del DHEA o quella indiretta, come precursore di estrogeni. Le nostre cellule non esprimono ERα, responsabile di molti
fenomeni di differenziamento, quindi si può escludere un suo coinvolgimento; non possiamo trascurare però altre vie d’azione degli estrogeni. Al momento non sono stati isolati recettori specifici per il DHEA, né di membrana né nucleari classici, tranne un recettore di membrana accoppiato a proteine G (43). Esperimenti recenti indicano che alcuni ormoni steroidei, oltre alla ben nota azione genomica, possono produrre rapidi effetti non genomici e modulare alcune proteine di membrana come canali ionici e complessi recettore-proteina G (43-46). Alcuni dati suggeriscono la possibilità che il DHEA possa agire modulando l’omeostasi del calcio intracellulare. Infatti il DHEA può modulare l’attività della Ca2+-ATPase della corteccia di ratto (47). Nei neuroni può anche comportarsi come modulatore allosterico del recettore NMDA (27), mentre in cellule muscolari il DHEAS influenza la depolarizzazione indotta da accumulo di calcio citoplasmatico (48). I gradienti di calcio, generati dalla stimolazione del recettore dell’NMDA, del GABA, o dei canali voltaggio-dipendenti, sono coinvolti nei riarrangiamenti cellulari che determinano fenomeni come la migrazione neuronale, la produzione dei neuriti e la creazione della complessa rete sinaptica (49-51). Il DHEA potrebbe quindi modulare l’ingresso di calcio e incrementare la crescita dei neuriti in cellule in via di differenziamento. A questo proposito è interessante considerare che il promoter del gene della MMP-9 contiene un elemento di risposta per il calcio (52). Una differente espressione di tale proteasi legata all’ingresso di calcio è stata osservata in cheratinociti e in cellule di carcinoma murino (53-55). Noi abbiamo dimostrato che il DHEA incrementa l’attività della MMP-9, è quindi ragionevole ipotizzare una induzione calcio-dipendente.
In conclusione i nostri dati mostrano un potenziamento dell’effetto differenziante dell’acido retinoico sul neuroblastoma ad opera del DHEA. Tale effetto non può essere spiegato con una regolazione a livello genomico, in quanto non coinvolge i recettori classici per gli ormoni steroidei; in alternativa si può ipotizzare una modulazione della concentrazione di calcio intracellulare, che potrà essere verificata in futuro con studi di imaging di calcio.